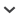In the 1970s, the basic techniques for DNA fingerprinting, Southern Blot analysis and Restriction Fragment Length Polymorphism (RFLP), were developed (Rudin, 2002). In 1985, Alec Jeffreys used RFLP as a way to identify individuals from their DNA, a process he termed DNA fingerprinting (Jeffreys et al., 1985). In 1985, Kary Mullis and colleagues from the Cetus Corporation first published the Polymerase Chain Reaction (PCR) method of replicating specific regions of DNA utilizing gene specific primers and specific thermocycler conditions (Saiki et al., 1985).
The combination of RFLP with PCR led to the possibility of identifying individual specific regions of DNA from limited samples, such as a single hair shaft with an intact follicle left at a crime scene. This technology, termed DNA profiling, would allow detectives to link a suspect to a crime scene by using his/her DNA fingerprint. The United Kingdom became the first country to use DNA profiling to exonerate one suspect of rape and to convict another for the same crime in 1987 (Canadian National DNA Database, 2003; Burns, 2005). In 1989, the United States (U.S.) had its first case overturned because of DNA evidence and the U.S. federal government began developing regulatory standards for DNA collection and handling procedures. In 1992, the National Research Council deemed DNA testing a reliable method to identify a criminal suspect, which prompted the technology to rapidly enter the mainstream court system. The Federal Bureau of Investigation (FBI) established the National DNA Index System, enabling city, county, state, and federal law enforcement agencies to compare DNA profiles electronically in 1998 (Burns, 2005). This software program is referred to as CODIS (COmbined DNA Index System) and contains DNA profiles from convicted offenders, missing persons, and unsolved crimes (FBI, 2006).
In 1994, DNA profiling was further advanced by performing PCR to evaluate specific loci that are variable between individuals, including monozygotic twins (NIJ, 2002). These regions are referred to as short tandem repeats (STRs) and the FBI uses a standard set of 13 of these for CODIS analysis and databanking (NIST, 2007). The simplicity of this assay is that the primers for PCR for the 13 loci can be mixed into one reaction, therefore giving rise to what is known as multiplex PCR. The specificity of these 13 loci lies in the fact that the odds that two individuals share the same DNA profile based on these loci is about one in one billion (NIJ, 2002).
Today DNA fingerprinting is used in a multitude of ways, including paternity testing, identifying animal versus human remains, rape cases, murder trials, historical cases, military dog tags, missing persons, and disease/health issues (Butler, 2001). Television programs, such as “CSI,” have pushed forensic science and DNA fingerprinting into every household. While doing this, the field has been glamorized, exaggerated, and oversimplified.
This experiment was designed to provide students, in a classroom laboratory setting, a hands-on demonstration of the steps used in DNA forensic analysis by performing DNA extraction, DNA fingerprinting, and statistical analyses of the data. The PCR parameters were determined by using control human DNA and first doing the reactions individually. Next, the four forward and reverse primers were mixed to make a multiplex primer mix and the parameters tested using control human DNA. Once the parameters were determined, the experimental DNA was extracted from pop cans, pop bottles, plastic spoons, cigarette butts, and cheek swabs and used in multiplex PCR reactions. The resulting PCR products were analyzed by gel electrophoresis and visualized utilizing a gel photodocumentation system. From the gel, the DNA fingerprint of the individual could be determined and analyzed.
Materials
used pop cans, pop bottles, spoons, and cigarette butts
BuccalAmp™ DNA Extraction Kit with QuickExtract™ DNA Extraction Solution and Catch-All™ Sample Collection Swabs (EPICENTRE®)
Microcon centrifugal filter devices (Amicon)
GoTaq® Green PCR Master Mix (Promega)
10X TBE (for 1 liter: 108 g Tris base, 55 g boric acid, 40 ml 0.5 M EDTA, pH 8.0) and 1X TBE buffer
0.9% saline solution (9 g NaCl in 1 L nanopure water)
500 μg/mL ethidium bromide (Fisher) – working concentration
TE buffer (10 mM Tris-Cl, pH 7.5; 1 mM EDTA)
5% Chelex solution (Sigma)
6X orange/blue loading dye (Promega)
90% and 70% ethanol
Phenol Chloroform Isoamyl alcohol (PCI; Fisher)
3 M sodium acetate
thermocycler
Mini-PROTEAN® II Gel Electrophoresis Unit and 4–15% Tris-HCl ready-made polyacrylamide gels (Bio-Rad)
1.5 ml microcentrifuge tubes
pipets and tips
vortex and centrifuge
heating blocks or water baths
sterile razor blade, forceps, 100 base pair ladder (Bio-Rad)
control human DNA
gel electrophoresis equipment
platform shaker
gel documentation system
primers (Vermaas & Rhoads, 2004; Invitrogen)
Methods
DNA Extraction from Pop Bottles, Pop Cans, Plastic Spoons & Cheek Cells
DNA extraction was done in a laminar flow hood to avoid contamination from other sources, including the experimenter. The manufacturer's protocol (EPICENTRE®) was followed for DNA extraction with the following modifications. A Catch-All Sample Collection Swab was dampened with 0.9% saline solution and the excess solution was squeezed out by pushing the swab against the inside of a 1.5 ml microcentrifuge tube. For the cheek sample, an individual swished 0.9% saline solution in his/her mouth for 10–15 seconds and spit the solution into a beaker to be discarded. The cheek, pop bottle, pop can, and plastic spoon were brushed with the swab at the locations the DNA was likely to be found. This was inside the cheek, at the mouth of the bottle, the lip of the can, or the bowl of the spoon. They were swabbed at least 20 times. The swab was dried at room temperature for 15 minutes and placed in a microfuge tube containing Quick Extract™ DNA Extraction Solution, rotated five times, and pushed against the inside of the microfuge tube to remove excess liquid. The microfuge tube was vortexed, placed in a heating block at 65° C for one minute, vortexed again, returned to the heating block at 98° C for two minutes, vortexed a third time, and stored at −20° C.
All DNA sample types (pop bottles, pop cans, plastic spoons, and cheek cells) were further purified by adding Phenol Chloroform Isoamyl alcohol (PCI) in a 1:1 ratio to each microfuge tube. PCI is highly toxic. Special care and handling should be taken when using it. Working in a fume hood is highly recommended. Each microfuge tube was vortexed and centrifuged at 13,000 rpm for two minutes. The top aqueous layer containing the DNA was carefully transferred into new microcentrifuge tubes and the bottom layer discarded. The DNA was precipitated by adding 3 M sodium acetate (NaAc) in a 1:10 ratio and 1 ml of 90% ethanol (EtOH) to each microfuge tube. The microfuge tubes were mixed by vortexing and placed at −20° C for approximately 24 hours. DNA samples were centrifuged at 16,000 rpm for 20 minutes at 4° C, the supernatant discarded, and 1 ml of 70% EtOH added to the pellet to wash off any excess salt. The DNA pellets were centrifuged for five minutes at 13,000 rpm, the supernatant discarded, and the DNA pellet dried. The DNA samples were placed with caps open in a 37° C incubator for 10–15 minutes, the DNA resuspended in 25 μl of TE buffer, and placed at −20° C for storage.
DNA Extraction from Cigarette Butts
DNA was extracted from cigarette butts following the protocol outlined in Hochmeister et al. (1991). In the laminar flow hood using a sterile razor blade and forceps, three cross-sections approximately 3 mm wide were cut from the filter end of a cigarette butt. The cuttings were placed in a microfuge tube with 1 ml of 5% Chelex solution and vortexed for 30 seconds. The microfuge tube was placed in a heating block at 56° C for 30 minutes, vortexed, boiled by heating to 100° C for eight minutes, vortexed again, and centrifuged at 13,000 rpm for five minutes to pellet the Chelex. The supernatant was carefully transferred to a centrifugal filter tube and centrifuged at 4,000 rpm. The retentate was washed with 2 ml of TE buffer and was stored at −20° C. The cigarette butt DNA was further purified by PCI extraction and precipitated with NaAc and EtOH as described previously.
PCR Amplification & Gel Electrophoresis
PCR reactions were prepared in the laminar flow hood with DNA extracted from U937 monocytic cells as a positive control, without DNA as a negative control, and with DNA from the cheek sample, cigarette butt, pop can, pop bottle, or plastic spoons. The positive control and negative control reactions were prepared with both the individual primer sets and with the multiplex primer mix containing all four primer sets. The experimental reactions were prepared with only the multiplex primer mix. These were all prepared by adding 12.5 μl GoTaq® Green Master Mix, 5.0 μl of U937 DNA (0.1 μg/μl; positive control), 5.0 μl of water (negative control) or 5.0 μl of experimental DNA, 2.0 μl of individual primers sets or multiplex primer mix (final concentration of each primer is 2.5 μM), and 5.5 μl of water for a final reaction volume of 25 μl. All reactions were vortexed, centrifuged, and placed in the thermocycler using the following parameters:
Initial denaturation: three minutes at 94° C
Denaturation: one minute at 94° C*
Annealing: one minute at 58° C*
Extension: three minutes at 72° C*
Repeating Steps 2–4(*): 30 cycles
Hold: 4° C (modified from Vermaas & Rhoads, 2004).
PCR was increased to 40 cycles for pop can and pop bottle DNA samples, and to 50 cycles for the cigarette butt DNA samples. The increase in cycle numbers for these DNA samples was due to the limited concentration of DNA recovered from these sources. The lower the concentration of DNA, the higher the number of cycles needed to achieve adequate amplification to observe a PCR product on the gel.
A four to fifteen percent Tris-HCl ready-made polyacrylamide gel was pre-run at 130 V with 1X TBE buffer for approximately 15 minutes. Marker DNA was made by adding 2.0 μl of 6X blue/orange loading dye, 5.0 μl TE buffer, and 3.0 μl of Bio-Rad 100 base pair ladder. Ten microliters of either PCR product or marker was loaded into the gel and electrophoresed at 100 V for approximately one hour at room temperature. After the bottom dye band had run through the gel, the gel was placed in ethidium bromide (final concentration = 0.5 μg/mL) diluted in water and placed on the shaker for 30 minutes. A picture was taken using a gel photodocumentation system for comparison and analysis of the gel.
Results
Each individual primer set was initially tested using control human DNA extracted from U937 monocytic cells (Figure 1). Any human genomic DNA would work, but U937 DNA was readily available in the lab. The gel shows:
a PCR product at ~320 bp for CSF1PO (Lane 2)
a ~390 bp product for Y-GATA-H4 (Lane 4)
two products at ~180 and ~190 bp for HUMTH01 (Lane 6)
a ~215 bp product for D7S820 (Lane 8).
Figure 1.
PCR performed with individual primer sets using U937 DNA. Lane 1 = 100 bp ladder; Lane 2 = CSF1PO + U937; Lane 3 = CSF1PO no-DNA control; Lane 4 = Y-GATA-H4 + U937; Lane 5 = Y-GATA-H4 no- DNA control; Lane 6 = HUMTH01 + U937; Lane 7 = HUMTH01 no-DNA control; Lane 8 = D7S820 + U937; Lane 9 = D7S820 no-DNA control. CSF1PO (Lane 2) shows a PCR product at ~320 bp (arrow), Y-GATA-H4 (Lane 4) shows a PCR product at ~ 390 bp (arrow), HUMTH01 (Lane 6) shows PCR products at ~180 and 190 bp (arrows), and D7S820 shows a PCR product at ~215 bp (arrow). Bands without arrows are nonspecific PCR products.

These product sizes are within the expected ranges for the loci tested (Table 1). No PCR products were seen in the no-DNA negative control lanes. Nonspecific PCR products were detected but were outside the expected range of sizes for the loci tested.
Table 1.
STR loci utilized in multiplex PCR in this study. All loci are described in Vermaas and Rhoads (2004) and NIST (2007). In addition, CSF1PO is described in Hammond et al. (1994), D7S820 is described in Jin et al. (1997), HUMTH01 is described in Edwards et al. (1991) and Hammond et al. (1994), and Y-GATA-H4 is described in White et al. (1999).

Once the individual primer sets were tested, the four primer sets were mixed together to perform a multiplex PCR. This was first done using control human U937 DNA (Figure 2). The gel shows successful amplification of three of the four primer sets at once. A ~390 bp product represents CSF1PO amplification, two products at ~180 and ~190 bp represent HUMTH01, and a ~215 bp product represents D7S820. Amplification for Y-GATA-H4 was not detected. The reaction was attempted multiple times, but each time Y-GATA-H4 was not detected. This suggests that Y-GATA-H4 primer annealing is being hindered by either one of the other primer sets or by some other factor in the PCR reaction. There were no bands present in the no-DNA negative controls as expected. The products for CSF1PO, HUMTH01, and D7S820 on the multiplex PCR gel (Figure 2) coincide with the same size products from the gel testing the individual primer sets (Figure 1). After positive amplification was seen with control DNA and three of the four primer sets in the multiplex primer set mix, the cigarette butt, the pop can, the pop bottle, the cheek cells, and the plastic spoon (Figure 3) were tested with the multiplex primer set mix.
Figure 2.
PCR performed with multiplexed primers using U937 DNA. Lane 1 = 100 bp ladder; Lane 2 = multiplexed primers + U937 DNA; Lane 3 = no DNA control. PCR products for CSF1PO (~320bp), D7S820 (~215bp), and HUMTH01 (~180 and 190bp) are marked with arrows. The ~390bp PCR product for Y-GATA-H4 was not apparent. The bands without arrows are nonspecific PCR products.

Figure 3.
Multiplex PCR for cigarette butt, spoon, pop bottle, pop can, and cheek DNA samples. Lane 1 = 100 bp ladder; Lane 2 = cigarette butt DNA (Subject 1); Lane 3 = bottle DNA (Subject 1); Lane 4 = can DNA (Subject 1); Lane 5 = spoon DNA (Subject 1); Lane 6 = cheek DNA (Subject 2); Lane 7 = negative control. C = CSF1PO PCR products; D = D7S820 PCR products; H = HUMTH01 PCR products. The bands without arrows are nonspecific PCR products.

The cigarette butt, pop can, pop bottle, and spoon PCR reactions all show one PCR product at ~325 bp for CSF1PO, one product at ~220 bp for D7S820, and two products at ~200 and ~185 bp for HUMTH01. For the cheek PCR reaction, two products at ~330 and ~320 bp are detected for CSF1PO, one product at ~230 bp for D7S820, and three products (tri-allelic expression) at ~200, ~180, and ~170 bp for HUMTH01. These product sizes are within the expected ranges for the loci tested (Table 1). No PCR products were seen in the no-DNA negative control lanes. Nonspecific PCR products were detected but were outside of the expected range of sizes for the loci tested.
The PCR products for the three loci are the same size for the cigarette butt, pop can, pop bottle, and spoon reactions, showing that the DNA originated from one individual, whereas the cheek DNA was extracted from a sample given by a different individual. The use of DNA samples from two different individuals was done to demonstrate that differences in banding patterns can be detected using the methodology described. In addition, the increase in cycle numbers for cigarette butt DNA compared to pop can, pop bottle, and spoon DNA from the same individual demonstrates that increasing the cycle numbers can increase the amplification of the target gene products. These PCR products appear brighter and more intense. In addition to Y-GATA-H4 amplification being inhibited in the multiplex primer set mix, we would not expect it to be amplified in these reactions as it is a male specific marker and all of the DNA samples were derived from female donors.
Discussion
Television today oversimplifies much of forensic science. With this experiment, students are exposed to the actual methods of DNA analysis used in forensics by performing DNA extraction, amplification by PCR, and gel electrophoresis in a laboratory. From this information, the students could compare “suspect” DNA to DNA found at the crime scene to determine the identity of the DNA fingerprint.
In this experiment, positive amplification was seen as bands on the gel when testing the individual primer sets and the multiplex primer mix with or without DNA in the reaction mix. This showed positive PCR amplification of target STRs, the specific area of DNA chosen to be amplified, in three of the four loci tested. The absence of PCR products in the no-DNA negative controls demonstrates that there was no amplification occurring in these reactions and that there was not DNA contamination of the reagents used in the PCR reactions. This verifies that amplification of the target gene sequences was specific and did not produce spurious PCR products. The approximate PCR product sizes for the individual primer sets coincided with the PCR products with the same size from the multiplex PCR when using the control DNA for all loci except for Y-GATA-H4. This suggests that more than one specific area of DNA can be successfully amplified at a time and that all of the amplification products can be individually visualized. The lack of amplification of Y-GATA-H4 does not negate this finding. It does suggest that a different male specific STR should be used if deemed necessary.
The multiplex gels with DNA extracted from a pop bottle, pop can, cigarette butt, plastic spoon, and cheek swab also show successful amplification of target STRs as seen by the presence of PCR products that fall within the expected size range for the loci as shown in Table 1. Since the PCR product sizes for CSF1PO, HUMTH01, and D7S820 for the cigarette butt, pop can, pop bottle, and spoon samples were estimated to be the same size, we can deduce that they are from one individual. This is in contrast to the PCR product sizes for these three loci from the cheek sample, which was from a different individual. The comparison shows that the multiplex PCR protocol can be used to distinguish between individuals.
DNA could be extracted from more individuals to further exemplify the differences in the DNA fingerprints. DNA as a molecular fingerprint for each individual could be discussed, as well as how DNA fingerprinting is utilized in different areas such as the criminal justice system, military dog tags, organ donor compatibility, and species genome projects. Since the DNA extracted in this experiment was primarily from saliva, DNA could also be extracted from other items such as blood, hair, or skin to show that an individual's DNA is the same regardless of the source. To differentiate between females and males, a different male specific STR marker could be used and put into the multiplex PCR primer set.
As a teaching experiment in a laboratory setting, a mock crime scene could be set up so that students could collect samples for DNA extraction from items such as blood on a shirt, the rim of a ball cap, or a used cigarette. The DNA could be extracted from these samples and each student could also extract his/her own DNA utilizing the cheek swab method. Amplification and gel electrophoresis could be done comparing the PCR products to determine if one of the students committed the crime. The activity teaches students how long the process of DNA fingerprinting takes. Connecting a DNA fingerprint to the perpetrator of a crime is not something that can be done in minutes or even hours, as portrayed on television. Many times in real-life crime scenes, there is no known match to the DNA tested and the crime becomes a “cold case” until a match in CODIS is made. This is not popular opinion based on television shows but, in reality, it is a common occurrence of which students should be aware. These are a few of the topics that could be discussed. If the instructor wants to incorporate probability testing into the laboratory exercise, the DNA profiles can be used to determine allele frequencies and to perform Hardy-Weinberg analysis. This laboratory exercise demonstrates how DNA fingerprinting is performed and how long it takes. It also shows that DNA fingerprinting is a useful tool in forensic science. Lastly, it debunks the myth portrayed in popular TV shows that obtaining DNA evidence is a guarantee that the crime will be solved in an hour.
Acknowledgments
We thank two anonymous reviewers and Darby Carlson for their helpful comments on improving this manuscript. This project was granted Institutional Review Board approval (IRB #071305-1) to develop the standard operating procedure to be used as a laboratory exercise. This project was supported by an undergraduate research grant from the University of Nebraska at Kearney (UNK) Undergraduate Research Council, the UNK Biology Department, and the UNK College of Natural and Social Sciences.